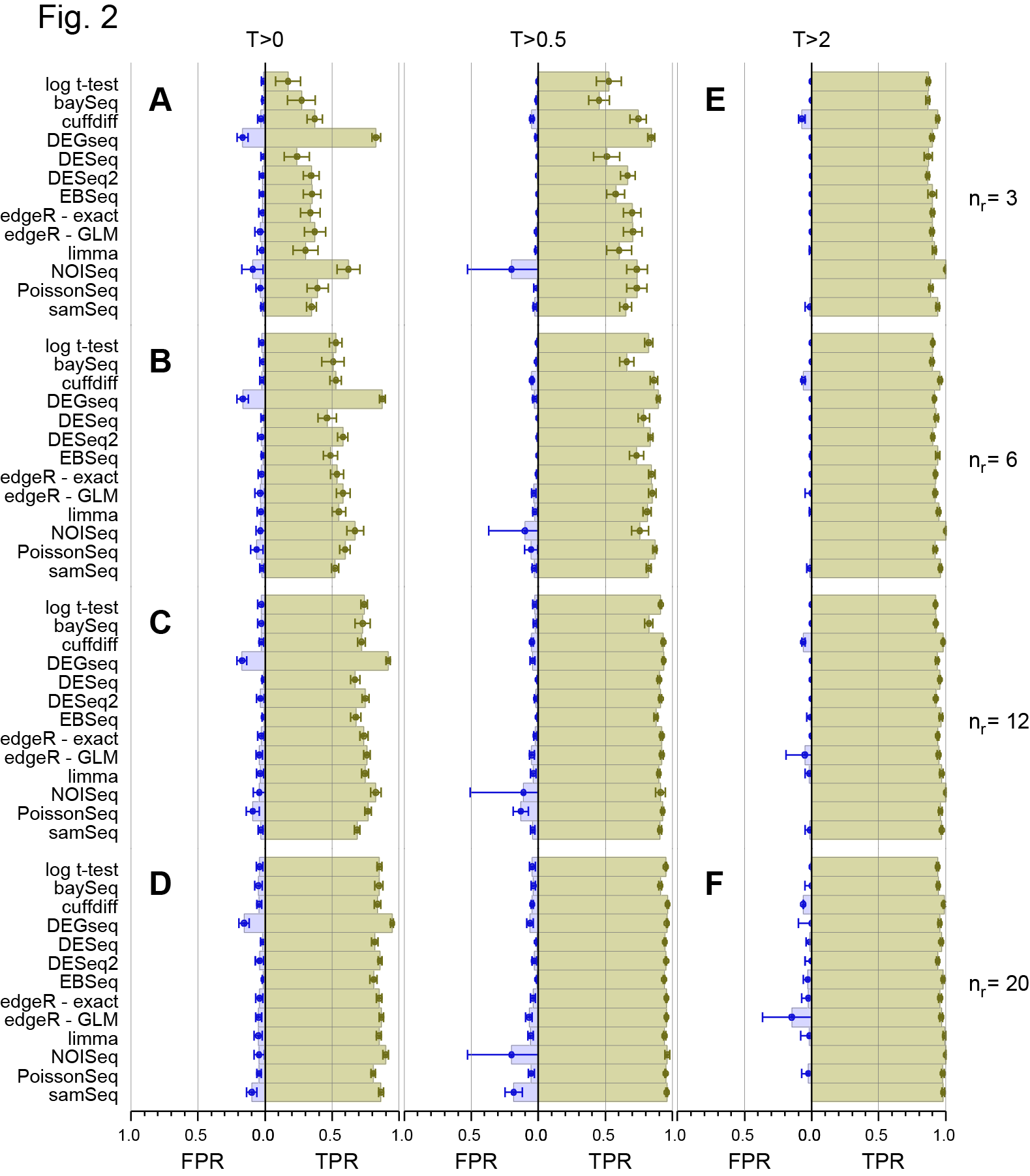
On 28th March, the main paper in our study of a 48 replicate RNA-seq experiment designed to evaluate RNA-seq Differential Gene Expression (DGE) methods was published on line by the journal RNA (Schurch et al, 2016, RNA). I’ve discussed this experiment in more detail in previous posts (48 Replicate RNA-seq experiment and 48 Rep RNA-Seq experiment: Part II ) so please see them for background information. Those who have been following the story on twitter since then will know that we made a mistake in a couple of the figure panels in that paper and this affected the conclusions about the False Discovery Rate (FDR) of one of the 11 methods we examined (DESeq2). Thanks to great help from Mike Love (the developer of DESeq2) and the fact we had made all the code and data available, we together identified the problem and have now corrected both the figures and the text in the manuscript to reflect the correct results. We caught this error early enough so that the online (and print !) versions of the manuscript will be corrected. Unfortunately, the revised version won’t be on-line until 16th May so I’m putting the two most important revised figures here to avoid confusion about the performance of DESeq2 which we now find is at least as good as DESeq and the other methods that performed well in our test.
Revised Figure 2: Schurch et al, 2016, RNA
Revised Figure 4: Schurch et al, 2016, RNA
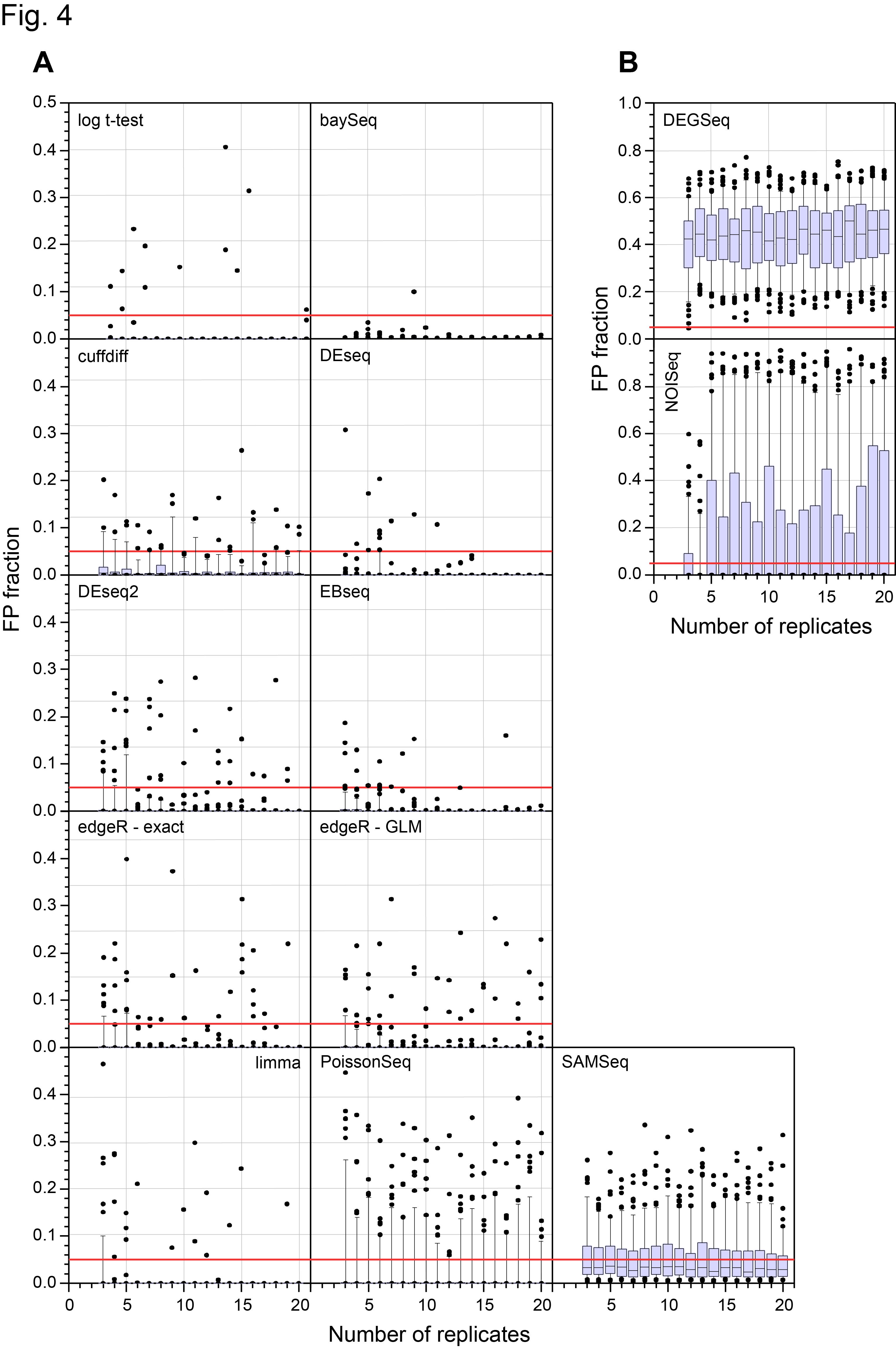
In case you were wondering, the mistake was caused by a combination of “features” in two packages we used, SQLite and perl PDL. DESeq2 by default pre-filters the data before calling differential expression. As a result it outputs floating point numbers and “NA”s. NA has a special meaning in the language R. We stored the output in an SQLite database which happily puts NAs and numbers in the same field. We then read the SQLite database with a perl program and the PDL library in order to make the plots. This silently converted NAs to zeros which is not what you want to happen! The effect was to call some genes as highly significant when in fact they were not called this way by DESeq2. This bug also influenced the results for DEGseq but did not affect our overall conclusions about that method. The bug has been fixed in the version of the code now on GitHub (https://github.com/bartongroup).
Although the team and I are embarrassed that we made a mistake when we had tried very hard to do this big study as carefully as possible, we’ve found the last few weeks a very positive experience! Mistakes often happen in complex research but they can go unnoticed for a long time. Here, the turn-round time was a few days on social media and a little over a month in the conventional literature. I think this illustrates how early and open access to manuscripts, code and data can help identify problems quickly and lead to better (or at least more reliable) science!
I’ll write in a separate post about the publication process and add my views on how I think it could be improved, but for now I just want to thank everyone involved in fixing this problem. Firstly, Mike Love who has been very professional throughout. He emailed as soon as he spotted the anomaly and then worked with us over a few days to identify whether it was real or an error. Thanks Mike! Secondly, the staff at the journal RNA, in particular the production editor Marie Cotter who responded very quickly and helpfully when we told her we needed to change something in the manuscript. Finally, the entire team in Dundee who worked on this project to find the bug and revise the m/s in good time. Special thanks to Nick Schurch who somehow managed to juggle his small children and other family commitments to make it all happen quickly!
How does this affect the conclusions?
The main effect is what I say in the post. DESeq2 now groups with the other “good” methods.
Thank you for following this up and clarifying the situation! I was already worried a bit!
No problem. The revised manuscript is now on the RNA journal website so you can see the full text and new figures in context. See: http://rnajournal.cshlp.org/content/22/6/839.full.pdf+html